
HEMOLYTIC ANEMIAS
- Premature destruction of erythrocytes (Er)
- ↑reticulocyte count, BR, LDH. ↓serum haptoglobin
- Divided into intrinsic (inherited) and extrinsic (acquired)
- 4 types of intrinsic haemolytic anemias
- Structural haemoglobinopathies – sickle cell disease synthesis of a structurally abnormal Hb protein
- Thalassemias – quantitative abnormality (decreased synthesis) of a globin chain
- Enzyme defects – G6PD-D, PK-D
- Membrane defects – hereditary spherocytosis
2. HEREDITARY SPHEROCYTOSIS (HS)
RBC Membrane
- 3 main components
- Phospholipid (PL) bilayer
- Integral membrane proteins and glycoprotein – embedded in the PL bilayer
- Important transmembrane proteins – band 3 protein and glycophorin
- Cytoskeleton scaffold – gives RBC its shape. Mainly composed of spectrin. Ankyrin binds spectrin to band 3 protein
Epidemiology
- MC inherited haemolytic anaemia in North Europe
- 75% are AD
Pathophysiology
- Defective vertical attachment between PL bilayer and the cytoskeleton scaffold
- Results from mutations in the gene for ankyrin
- Defective vertical attachment causes loss of PLs from the cell membrane
- Surface area of RBC decreases and cell assumes shape of a sphere
- Spherocytes are less flexible than normal RBCs and get trapped and destroyed in the spleen
- Intrinsic defect with extravascular hemolysis
- Increased permeability to Na+ and K+ means the pump is constantly running, which causes additional metabolic stress
- Cells have an increased requirement for glucose
Clinical features
- Highly variable
- Neonatal hyperbilirubinemia
- Older patients have mild/mod anaemia with hyperbilirubinemia and mild splenomegaly
- Bilirubin gallstones
- Pts can have exacerbations of anaemia associated with infections
Diagnosis
- MCV is normal/low. Increase MCHC – specific for this haemolytic anaemia
- Blood smear shows microspherocytes
- ↑reticulocyte count
- DAT should be done to exclude immune haemolytic anaemia
- Osmotic fragility test – classic test for HS
- Er incubated in saline solutions with osmolality ranging from normal to pure water
- Percent hemolysis is measured by spectrophotometer
- Er from HS patients hemolyse at higher saline concentration than normal cells
Treatment
- Splenectomy, folic acid, transfusion
- Should be delayed until 3-5 years of age due to risk of OPSI
3. ENZYME DEFECTS
Glucose-6-Phosphate Dehydrogenase Deficiency (G6PD-D)
Epidemiology
- One of the MC genetic diseases in the world
- XR – men develop disease, but women are usually asymptomatic carriers
- MC in Africa, Mediterranean, Asia
Pathophysiology
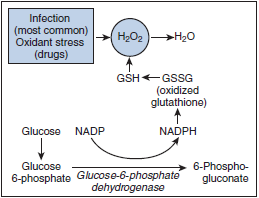
- First enzyme in hexose monophosphate shunt, which is required to generate NADPH, which is needed for regeneration of glutathione by glutathione reductase
- In absence of sufficient glutathione, Hb is oxidised and precipitates in the cells (Heinz bodies) – results in hemolysis
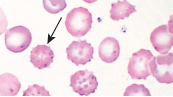
- Aggregates of oxidised Hb are removed from the cell by the spleen, resulting in bite cells
- Level of G6PD is highest in reticulocytes and lower in aged cells
- In normal people the activity of the enzyme remains enough to protect older cells from oxidative stress
- Normal G6PD half life is 62 days
- In the common African variant of G6PD deficiency half life is 13 days
Clinical features
- Most patients are not anaemic and have no hemolysis at baseline state
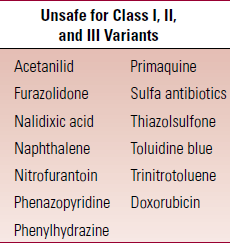
- African variant is Class III – episodes of hemolysis are precipated by infection, oxidative drugs, chemicals, surgery
- Episodes of hemolysis are indicated by sudden onset jaundice, pallor, dark urine, abdominal pain
- Mediterranean variant (class II) is MC in Caucasians
- Uncooked fava beans are common cause of hemolysis in these patients
- Class I variants are very unstable
- Anemia and jaundice noted in neonates
Diagnosis
- Bite cells in peripheral smear (pic)
- Fluorescent screening test for NADPH production
- DAT –ve
Treatment
- Avoid conditions that predispose to hemolysis
- Treat infections promptly
- Transfusions for infants with marked hyperBR

 Pyruvate kinase deficiency (AR)
Pyruvate kinase deficiency (AR)
Epidemiology
- MC in pts of North European and Mediterranean descent
Pathophysiology
- Defect in Emden-Meyerhof pathway
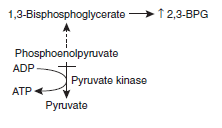
- Er with PK deficiency generate less ATP and NADH from glucose
- 2,3-BPG accumulates in RBCs
- Since ↑2,3-BPG facilities oxygen unloading, patients tolerate anaemia well and are asymptomatic despite ↓Hb ( Right shift of Hb-O2 dissociation curve >> ↑release of O2 to tissues)
- Decreased ATP leads to cellular dehydration and formation of echinocytes
Clinical features
- Neonatal hyperBR
- Older children and adults – chronic hemolysis
- Splenomegaly
- Infections, surgery, pregnancy can precipate acute exacerbation of hemolysis
 Aplastic crisis can occur due to infx with parvovirus B19
Aplastic crisis can occur due to infx with parvovirus B19
Diagnosis
- enzyme assays show low PK activity
- Blood smear shows echinocytes (pic)
- PKLR gene mutation
Treatment
- Transfusions for neonatal hyperBR
- Older patients normally tolerate anaemia well so don’t require treatment
- splenectomy