
HEMOGLOBINOSIS/HEMOGLOBINOPATHIES – Sickle Cell (SCA)
- Mutations in the gene for a globin chain, resulting in synthesis of structurally abnormal Hb
- Most are β-chain mutations
- Inherited in an autosomal fashion – mainly AR (homozygotes)
- Heterozygotes are either asymptomatic or have mild disease
- Some Hb-pathies can interact – e.g. a person heterozygous for both HbS + HbC has clinical disease
- Whereas someone who is heterozygous for either one alone is asymptomatic
- Hb-pathies can also interact with thalassemias – someone who is heterozygous for both B-Thal and HbS will have clinical disease
- However, inheriting a gene for a-thal tends to decrease severity of HbS
- Hb-pathies can lead to haemolytic anaemia or ↑/↓ oxygen affinity
Terminology
Genotype – based on the specific globin chains that are present
- Heterozygous sickle cell (trait) – α2ββs (two normal a chains, one normal b chain, one b chain with sickle mutation)
- Homozygous sickle cell (anemia) – α2β2S (two normal a chains, two b chains with sickle mutation)
- Compound heterozygosity for HbS + HbC – α2βSβC
Phenotype – based on the haemoglobin types that are present (Hb in highest conc is written first)
- Heterozygous sickle cell (trait) – HbAS
- Homozygous sickle cell (anemia) – HbSS
- Compound heterozygosity for HbS + HbC – HbSC
- Patient heterozygous for both HbS and B-Thal – HbSA
Diagnosis of Hb-pathies
- Electrophoresis – separates Hb based on different size and electrical charges
- Performed on cellulose acetate
- Patients should not have been transfused for at least 90 days before ordering electrophoresis test
- Can be difficult to interpret in neonates due to physiologic elevation of HbF
 Other tests – sickle solubility test, isopropanol test
Other tests – sickle solubility test, isopropanol test
Sickle Cell Anemia (HbS)
Epidemiology
- MC in Africa. Also in areas of Turkey, Mediterranean, India
- Areas endemic in P.falciparum malaria
Pathophysiology
- Substitution of valine for glutamic acid at 6th Amino Acid position – β6GLU→VAL
- Deoxygenated HbS polymerises into long rigid structures which distort the cell into the sickle shape
- Anything that causes deoxygenation of Hb predisposes to sickling – e.g. hypoxia, acidosis, fever
- Initial sickling is reversible but repeated cycles of sicking and unsickling damage the cell
- Eventually RBCs become irreversibly sickled
- These rigid sickled cells obstruct small blood vessels, causing tissue infarctions (MC in spleen, BM, kidney, mesenteric and pulmonary vessels)
- They are also ‘sticky’ – adhere to endothelial cells and predispose to thrombosis
Clinical features
- Heterozygous HbS – Sickle cell trait (HbAS/α2ββS)
- Asymptomatic, normal Hb and CBC
- Microscopic hematuria – due to infarction of renal medulla (the hypoxic and acidotic environment causes even heterozygous cells to sickle)
- Homozygous HbS – Sickle cell anaemia (HbSS/ α2β2S)
- Children become symptomatic after 3 months of age (before that they are protected by high levels of HbF)
- Other sickle cell diseases
- HbSC – less severe than HbSS; retinopathy, pregnancy complications, mild splenomegaly
- HbSA – mild disease
Complications
- Vaso-occlusive crises
- Occlusion of small vessels and infarction of tissues. Pain in abdomen, bones, joints, muscles
- Sequestration crises
- MC in 3-4 years of age. Spleen suddenly becomes enlarged and engorged with blood. Can sequester a large portion of total blood volume, can be fatal
- Aplastic crises
- Occurs as a complication of infections – MC is parvovirus B19 infection which transiently halts the production of RBCs
- In patients with SCA, RBC survival is 10-20 days instead of 120 days so Hb drops more quickly
- Can be fatal without a transfusion
- Infections – MCC of death in SCD
- Children are maintained on penicillin prophylaxis against pneumococcal sepsis
- Acute chest syndrome
- Causes – infection, fat embolism, PE, vascular occlusions
- Findings – pulm infiltrates on CXR, chest pain, fever, hypoxemia, tachypnea, cough, dyspnea
- Splenic infarcts/altered splenic function
 Renal disease – infarction of medulla (see above)
Renal disease – infarction of medulla (see above)- Bilirubin gallstones – due to increased Hb turnover
- Leg ulcers
- Retinopathy
- Pregnancy complications
Diagnosis
- Sickle cell trait – normal CBC and smear
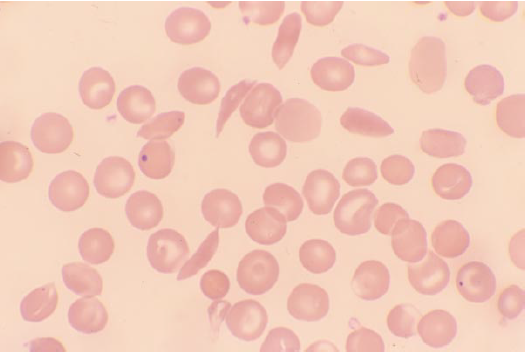
- SCA – ↓Hb (5-8g/DL), normocytic, target cells, sickled er (top smear)
- Howell-Jolly bodies – due to splenic infarction
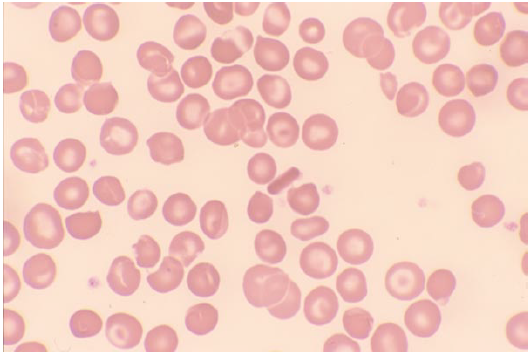 HbSC – rectangular HbC crystals (bottom smear)
HbSC – rectangular HbC crystals (bottom smear)- HbS + B-Thal – target cells and microcytosis
- Sickle solubility test – deoxygenated HbS has decreased solubility
- Cant differentiate between trait and SCA
- Can give false positive in presence of high HbF, so not a reliable test in infants
- Electrophoresis
- Performed on cellulose acetate at alkaline pH
- Useful in infants
- Can also distinguish between trait and SCA
Treatment
- Supportive – folate supplements, prophylactic penicillin
- Children vaccinated against S.pneumoniae, H.influenza, N.meningitidis
- Painful vaso-occlusive crises – oral hydration and analgesia
- Transfusions – for cerebrovascular accidents, acute chest syndrome
- Aim to maintain HbS <30%
- SE – Fe overload, alloimmunisation, haemolytic reactions, infections
- Prompt antibiotics for infections
- Acute chest syn – oxygen, empiric ABs, analgesia, transfusions
- Hydroxyurea – decreases painful crises, hospitalisations, episodes of ACS, increases HbF
- SE – myelosuppression
- BM transplant
- Genetic counselling
HbC
- MC in Africa
- Substitution of lysine for glutamic acid at 6th AA. HbC polymerises into crystals when deoxygenated
- Both heterozygous HbC (trait) and homozygous HbC (HbCC) are asymptomatic, no treatment required