
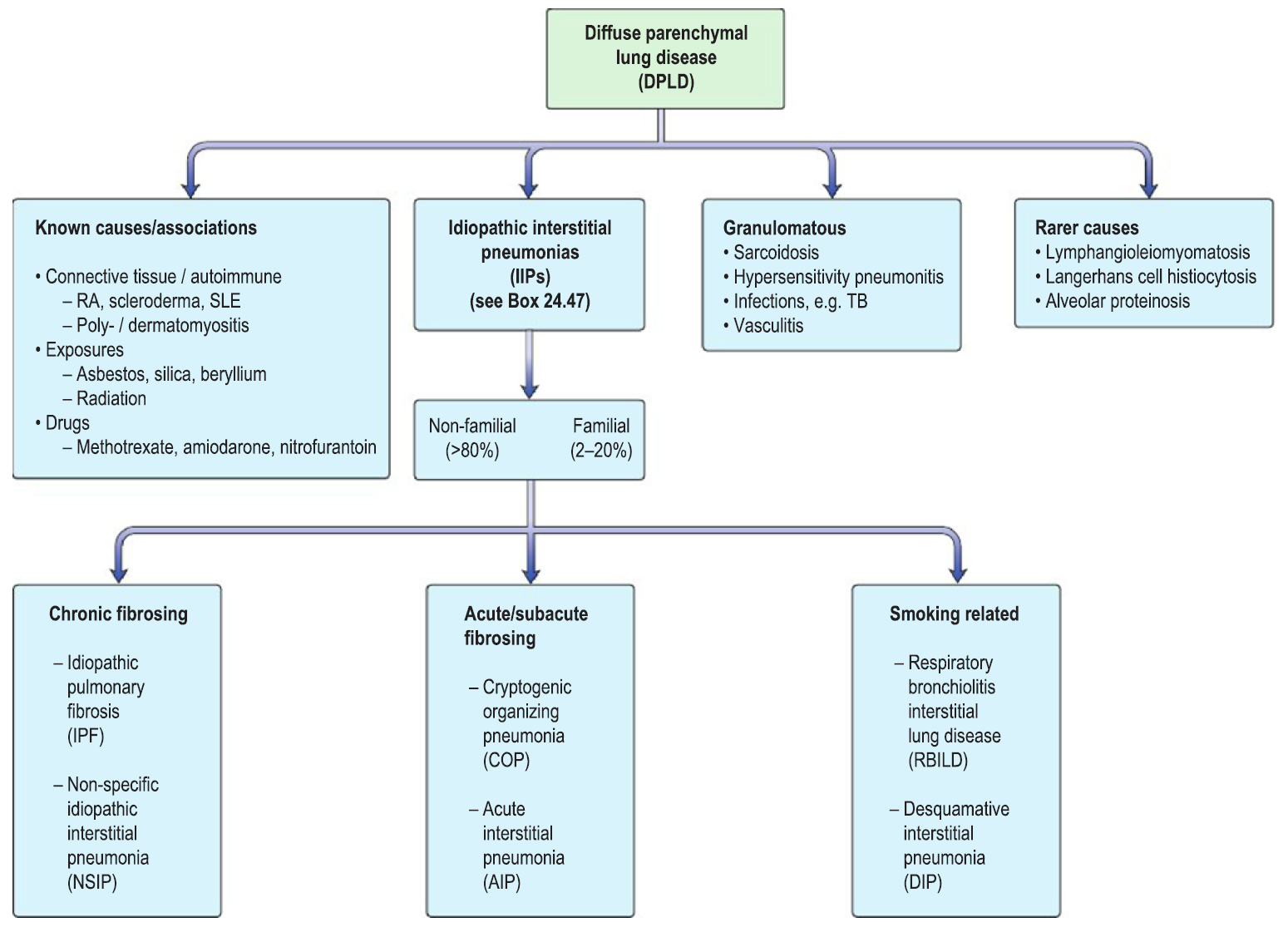
- Diffuse parenchyma lung diseases (DPLD) – heterogeneous group of conditions affected the pulmonary parenchyma/interstitium
- Often present with progressive dry cough and breathlessness
- IPF is defined as a progressive fibrosing interstitial pneumonia of unknown cause
Epidemiology/Etiology
- IPF is the most common IIPs (idiopathic interstitial pneumonias)
- Onset is MC in 60s
- MC in men
- Etiology unknown
- Risk factors – cigarettes, metal/wood/coal dust, silica, mold spores, EBV, methotrexate, chronic GERD
Pathogenesis
- Repetitive injury to alveolar epithelium due to an unknown stimuli
- Leads to uncontrolled wound healing mechanisms
- Overproduction of fibroblasts
- ↑deposition of extracellular matrix in the interstitium
- Structural integrity of lung parenchyma is disrupted
- Loss of elasticity
- Impaired gas exchange
Pathology
- Heterogenous appearance – areas of normal lung punctuated by areas of marked fibrosis
- Honeycomb cysts –enlarged air spaces with thick fibrotic walls (often filled with mucin and inflammatory cells)
- MC in subpleural areas
- Honeycomb cysts –enlarged air spaces with thick fibrotic walls (often filled with mucin and inflammatory cells)
Clinical features
- Insidious onset of progressive dyspnoea accompanied by dry cough ± sputum
- Finger clubbing
- Crackles
- Weight loss
- Fatigue
- Overtime disease can progress to cause pulmonary hypertension, cor pulmonale, type 1 respiratory failure
Diagnosis
- Respiratory function tests – show restrictive pattern
- FEV/FVC >70%
- Blood tests – ANA , RF
- CXR – ↑reticular shadowing, ground glass appearance
- CT – imaging of choice. Shows characteristic abnormalities
- Basal distribution
- Subpleural reticulation
- Traction bronchiectasis
- Honeycombing
Treatment
- Poor prognosis – median survival time is 2-5 years
- Smoking cessation
- Treat underlying GERD
- Immunosuppression – corticosteroids, azathioprine, cyclophosphamide
- Pirfenidone – antifibrotic agent
- Lung transplant